
The title of this post comes from an article published in a special virtual issue on the theme “Physical Organic Chemistry: Never Out of Style“[cite]10.1021/acs.joc.5c00426[/cite] There, Paul Rablen presents the case that the amount of o (ortho) product in electrophilic substitution of a phenyl ring bearing an EWG (electron withdrawing group) is often large enough to merit changing the long held rule-of-thumb for EWGs from being just meta directors into these substituents are best understood as ortho, meta-directors, with a preference for meta. I cannot help but add here a citation[cite]10.1039/CT8875100258[/cite] to the earliest publication I can find showing tables of both o,p and m-directing groups and dating from 1887, so this rule is 138 years old (at least).
Here I thought I might show some computational models (ωB97XD/Def2-QZVPP/SCRF=Dichloromethane)[cite]10.14469/hpc/15341[/cite] derived from the relative stability of the Wheland or σ-complex produced by protonating the Ph-EWG molecule in the three possible positions on the ring – and now taking the opportunity to add some unusual EWGs to the table to explore how far this effect might be pushed.

I start by looking at the results reported for benzonitrile (EWG = CN), for typical product distributions:
- o– (~16%), m– (~82%) and p– (~2%) are cited for nitronium ion as electrophile
- o– (23%), m– ( 74%) and p– (3% ) for chlorination
- o– (34%), m– (55%) and p– (1%) for uncatalysed bromination (see [cite]10.1002/jcc.23985[/cite] for an unexpectedly complex mechanism and kinetic analysis of this particular reaction)
- σ-complex calculations [cite]10.1002/poc.4457[/cite] which result in values of o– (43%), m– (55%) and p– (2%) for benzonitrile.
- The observation was made[cite]10.1002/poc.4457[/cite] that inclusion of a solvation correction substantially improved the agreement with the limited experimental information available to us regarding product distributions in EAS and the results below certainly confirm that (especially for benzonitrile). Solvent also has a significant effect on the optimised geometry of each system (see Table).
The calculations reported here[cite]10.14469/hpc/15341[/cite] are similar to those reported using a slightly different model[cite]10.1002/poc.4457[/cite]. For the specific example of benzonitrile, the authors of the original report expressed surprise that their computations showed that “the ortho and meta σ-complexes were … about equally stable“. The results for this blog show a slightly larger and perhaps more realistic (?) discrimination in favour of meta by 0.51 kcal/mol in the free energy.
Other noteworthy observations include that
- compared with CN, the iso-electronic isonitrile group NC is a strong and conventional o/p director, with a preference for p.
- The EWG R=BO (a known, albeit very unstable molecule[cite]10.1021/jo401942z[/cite]) is the next isoelectronic isomer of CN and it now reveals a very strong preference for meta-substitution, with only 3.5% ortho. So this group does NOT follow the proposed new rule of “ortho, meta-directors, with a preference for meta” although this is unlikely to ever be able to be tested experimentally due to the instability of this species (it readily trimerises).
- Finally in this isoelectronic progression for R=BeF, the calculations seem now to show that this is a strong o– director (61%) and that m is only 29%, again not following the newly modified rule but probably untestable.
- R=NO however does seem to be an example of the new modified rule, since the percentage of o– is as high as 23.8%. Here it is significant that for both the o– and m– σ-complexes, the NO group was calculated as being co-planar with the phenyl ring, thus indicating significant conjugation – but the p-isomer (2.3%) was twisted and hence un-conjugated (dihedral values shown below).
- The same result is obtained for R=NO2, with the p-isomer having a twist angle of 67°.
| Cationic intermediates in electrophilic substitution of Ph-R | ||||||
|---|---|---|---|---|---|---|
| R | ΔΔG298, kcal/mol (pop, %) ortho, |
rC-R Å |
ΔΔG298, (pop, %) meta |
rC-R | ΔΔG298, (pop, %) para |
rC-R |
| NC, gas | -4.72 (21.42) |
1.349 | 0.0 (0.01) |
1.369 | -5.51 (78.57) |
1.348 |
| NC, DCM | -2.50 (35.51) |
1.359 | 0.0 (0.56) |
1.377 | -2.86 (63.93) |
1.359 |
| CN, gas | -1.38 (60.56) |
1.423 | 0.0 (6.07) |
1.433 | +0.36 (33.37) |
1.425 |
| CN, DCM | +0.51 (27.68) |
1.428 | 0.0 (64.05) |
1.435 | +1.23 (8.27) |
1.433 |
| BO, gas | +0.96 (16.76) |
1.541 | 0.0 (82.34) |
1.540 | +2.72 (0.09) |
1.549 |
| BO, DCM | +1.99 (3.52) |
1.537 | 0.0 (96.34) |
1.532 | +3.93 (0.14) |
1.547 |
| BeF, gas | +0.23 (38.78) |
1.727 | 0.0 (56.73) |
1.714 | +1.53 (4.49) |
1.737 |
| BeF, DCM | -0.46 (61.21) |
1.748 | 0.0 (28.66) |
1.731 | +0.63 (10.13) |
1.762 |
|
|
||||||
| CF3, gas | +0.25 (30.86) |
1.524 | 0.0 (46.87) |
1.521 | +0.45 (22.27) |
1.533 |
| CF3, DCM | +1.45 (8.11) |
1.518 | 0.0 (89.66) |
1.513 | +2.22 (2.23) |
1.528 |
| NO, gas | +0.44 (25.07) |
1.460 | 0.0 (52.32) |
1.477 | +0.51 .22.61) |
1.395 |
| NO, DCM | +0.68 (23.84) |
1.458 | 0.0 (73.87) |
1.456 | +2.09 (2.29) |
1.429 |
| NO2, gas | +1.08 (13.38) |
1.487 | 0.0 (79.88) |
1.487 | +1.49 (6.73) |
1.476 |
| NO2, DCM | +1.80 (4.73) |
1.480 | 0.0 (94.25) |
1.478 | +2.73 (1.01) |
1.481 |
On to the suggested explanation,[cite]10.1021/acs.joc.5c00426[/cite] where interaction of the π-electrons from the σ-complex with the π* orbital from the EWG was suggested to be stronger not only for the m-isomer but also the o-isomer as compared to the p-isomer. This can now be quantified using NBO7 analysis, which indicates the energy of interaction between pairs of filled donor and empty acceptor orbitals.
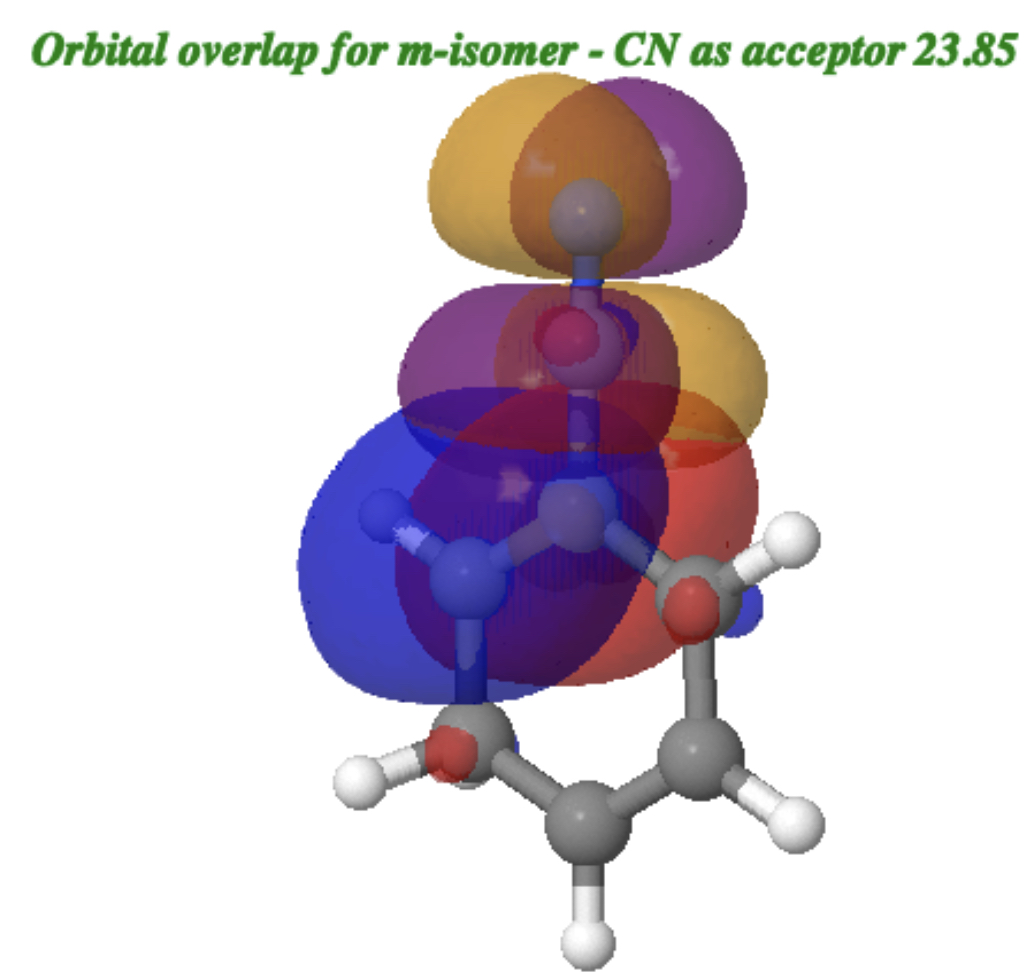
For the m-isomer[cite]10.14469/hpc/15354[/cite] of protonated benzonitrile, the overlap of the two orbitals (CN acting as an acceptor and the phenyl ring as a donor) is shown below (click on the image to get a rotatable 3D model) with blue positively overlapping with purple and red with orange. The NBO E(2) interaction energy is 23.85 kcal/mol (green bond above interacting with R=CN π*).
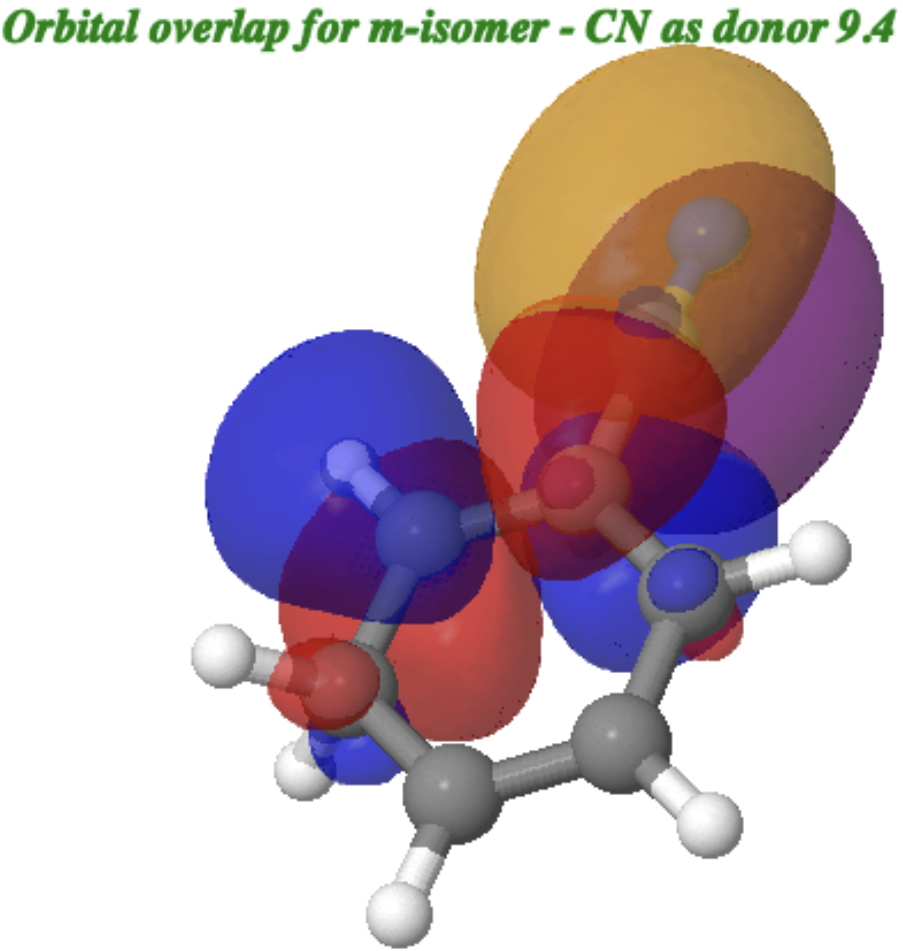
A reverse donation, from the π-system of the CN group now acting as a donor to the π* of the benzene ring acting as an acceptor has a smaller E(2) = 9.4kcal/mol. This shows that CN can act as both a donor and as an acceptor, but the latter effect is stronger.
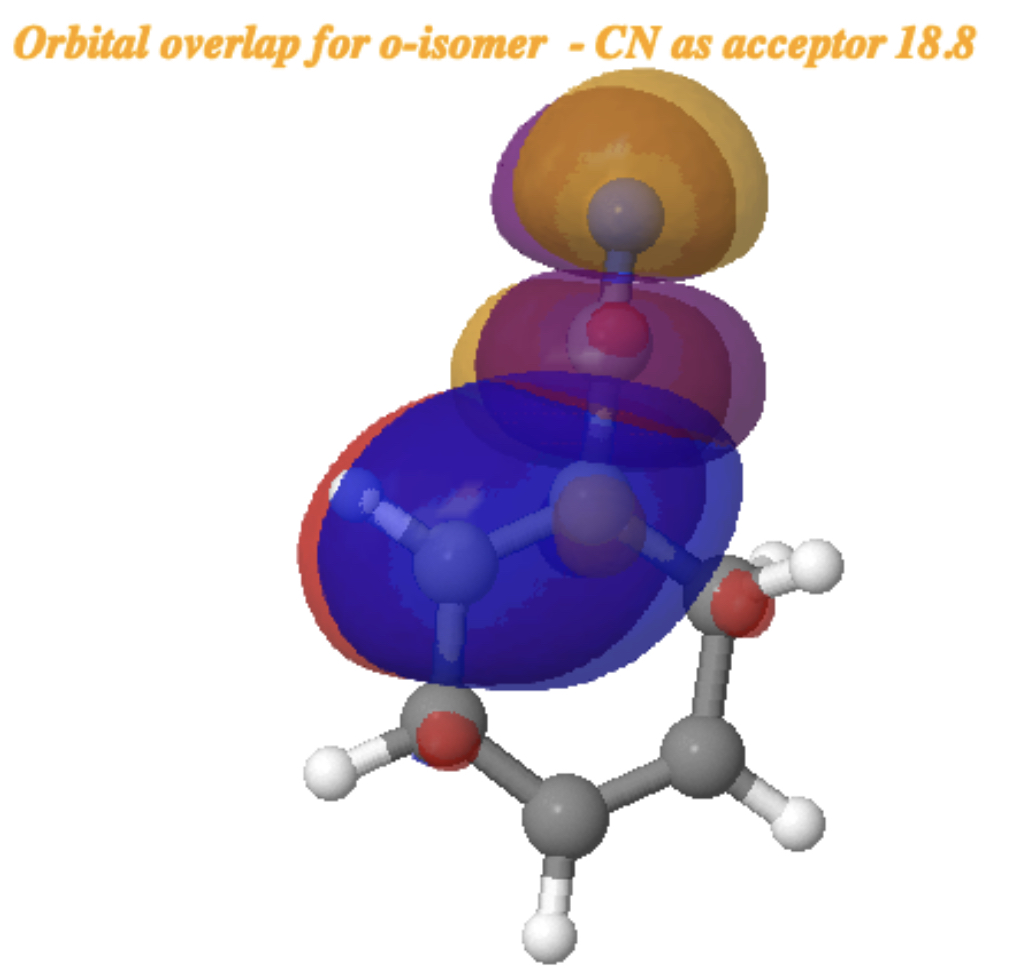
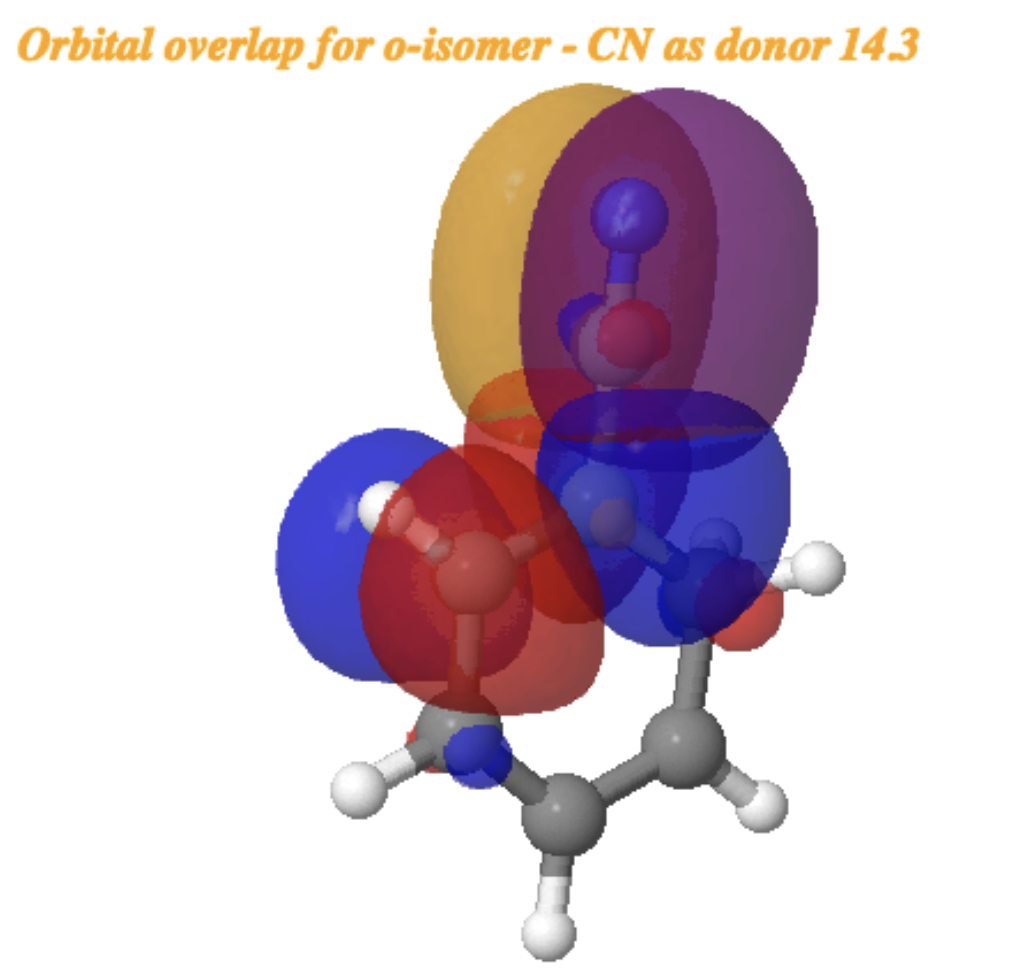
For the o-isomer[cite]10.14469/hpc/15355[/cite] (below), the NBO E(2) interaction energy is somewhat reduced to 18.8 kcal/mol (orange bond above interacting with R=CN π*). but is still considerable and more or less commensurate with the relative free energies of the o– and m-isomers.
A reverse donation, from the π-system of the CN group now acting as a donor to the π* of the benzene ring acting as an acceptor has a smaller E(2) = 14.3 kcal/mol. This again shows that CN can act as both a donor and as an acceptor with the latter effect the stronger.
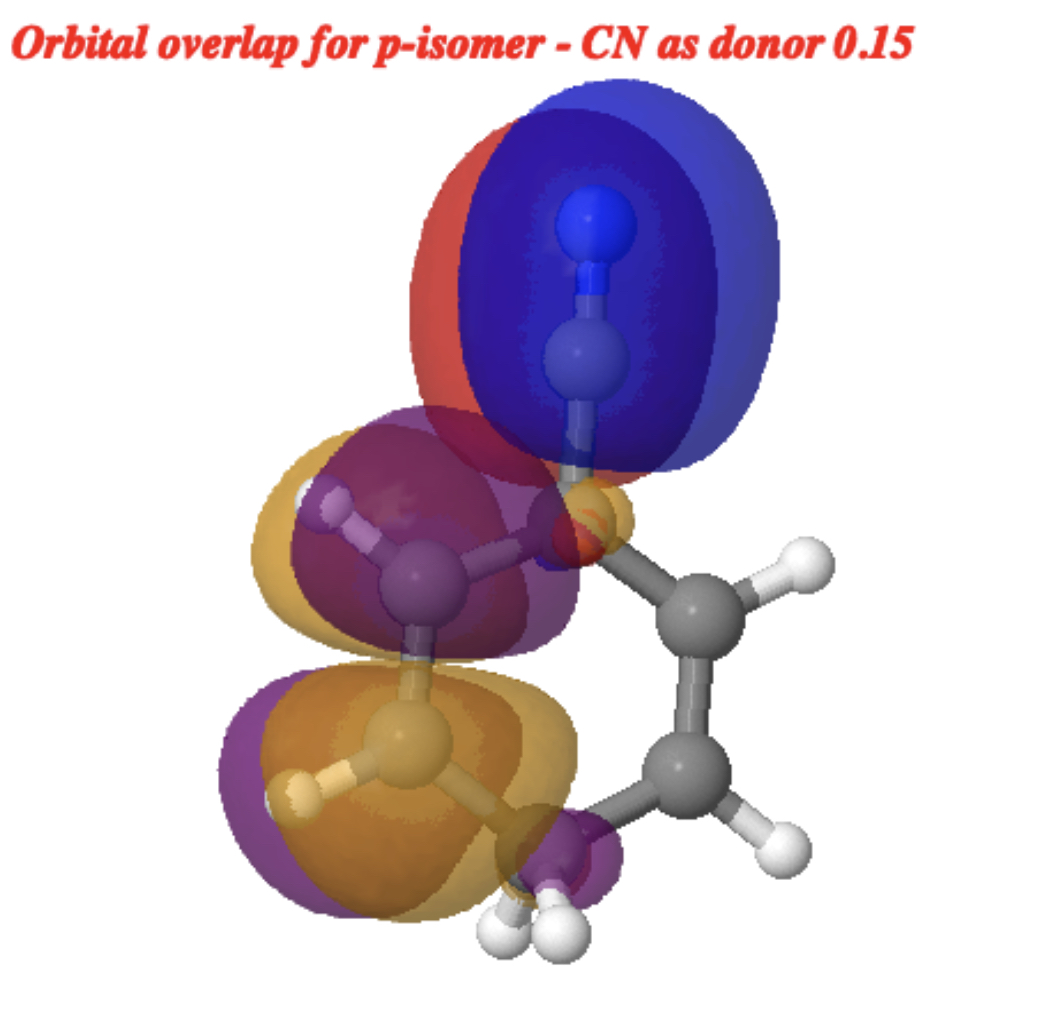
Things are quite different for the p-isomer[cite]10.14469/hpc/15353[/cite]. The equivalent CN-acceptor/phenyl-donor orbitals are shown below; they has no real overlap and the associated value for E(2) of 0.23 kcal/mol (red bond above interacting with R=CN π*) is tiny compared to that for the o- and m– isomers.

The reverse donation from the π-system of the CN group now acting as a donor to the π* of the benzene ring acting as an acceptor is equally small, E(2) 0.15 kcal/mol.
Furthermore, the p-isomer NBO E(2) interaction energy for the same atoms as with o– and m– shows two instances of 3.0 kcal/mol (because of the C2v symmetry), also very much reduced from 23.85 or 18.8 kcal/mol.
Although many other interactions can be found in the NBO analysis, this accounts for by far the largest difference between the o, m, and p isomers. These results also match with the observation made above that for R=NO, the o– and m-isomers are fully coplanar, but for the p-isomer the NO group is twisted by about 90° with respect to the phenyl ring. This is also reflected in the calculated torsional or twisting vibrations of the R group, being 89 cm-1 for m-Nitroso vs 23 cm-1 for o-nitroso and again 55 cm-1 for m-nitro vs 38 cm-1 for o-nitro.
So this new NBO7 orbital overlap analysis helps to quantify these effects (the reported qualitative analysis[cite]10.1021/acs.joc.5c00426[/cite] was based on molecular orbitals rather than localised NBO orbitals) and confirms that for some EWG groups at least, the o-isomer is almost as favoured as the m-form. Well, an observation that is 138 years old gets new light shone on it!
Related
You can leave a response, or trackback from your own site.