
Substituting a deuterium isotope (2H) for a normal protium hydrogen isotope can slow the rate of a chemical reaction if this atom is involved in the reaction mode. The magnitude of the effect, referred to as a kinetic isotope effect or KIE is normally 2-7, but higher values of 20 or even more♥ are sometimes observed due to a phenomenon known as proton tunnelling. So a recent report[1] of a 1H/2H of ~2440 for the following palladium catalysed reaction caught my eye:

When the protium in the solvent methanol and the hydrogen gas were replaced by deuterium, the rate of the reaction slowed by ~2400. This immediately begs one question: what was the % of deuterium incorporated into the 2H2 and MeOD? It would have to be >99.994% to eliminate any contribution from the presumably faster reacting 1H isotope, and this level of deuteriation is some ask! Leaving this issue aside, the authors then carried out some DFT modelling to come up with a proposed mechanism (below), which they refer to as a concerted triple hydrogen transfer reaction (the curly arrows by the way are mine; arrows are shown in the graphical abstract for this article but they are likely not curly electron arrows but simply schematic). The large value of the KIE was then attributed in part to a novel form of triple hydrogen tunnelling.

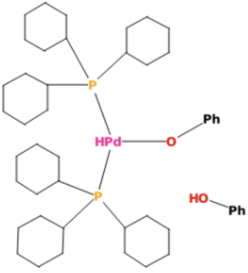
My second reality check was to search the crystal structure database for instances of the proposed catalyst containing a Pd-H substructure. Nine examples of compounds with such Pd-H bonds emerge, but none have the H-Pd(OR)3 motif shown above, which is likely to be a transient catalytic species rather than a stable isolable one. This species (FOTBAR)[2] with one OPh and two P ligands on the Pd is the closest match; although the trans relationship of the Pd-H and Pd-O bonds might preclude it functioning as a catalyst according to the mechanism above.
My next check related to the DFT procedure used, which was reported as B3LYP with apparently a 6-311+G(d,p) basis set, but no dispersion correction added. We had previously observed[3] that functionals such as B3LYP are not particularly well suited for transition metal modelling, preferring a newer variety such as MN15L.[4]
Finally, we also recollected our experience in modelling KIE effects using relatively modest basis sets such as 6-31G(d,p) and 6-311+G(d,p)[5] where we showed that the calculated KIE were inaccurate. Basis sets of eg Def-TZVPP or better were found to be essential. So here I test this hypothesis for a small selection of functional and basis sets as an initial exploration.
The calculations are published here (Table below).[6] Row 1 shows the values given in the article,[1] and for which a free energy of activation of 27.0 kcal/mol was indicated. Attempting to replicate this here, the main article declares that a 6-31G* basis set was adopted for the H, C, and O atoms… and the LANL2DZ basis set was adopted for Pd atoms. The supporting information records this instead as 6-311+G(d,p) for these atoms (Table S5. — B3LYP/6-311+G(d,p)/Lanl2dz level) which was used here. The basis used for Ti was not noted in either article or ESI; here it was set to 6-31G(d,p).[7] Using the Gaussian 16 program, my calculation gave the results shown in row 2, with geometry optimisation starting from the coordinates given in the ESI, giving a final RMS force of 0.000008 au – this value is not available for comparison with the original article, nor is the final total energy of the system. The imaginary transition state mode is 940 cm-1 compared to the reported value of 1306 cm-1, a not insignificant difference and which may arise from the reported basis set uncertainty. The bond lengths also differ somewhat, but the angle subtended at the Pd-H-C system is more or less linear. The newly computed free energy of activation is significantly lower. Re-modelling, but now including the effects of a methanol solvent also induces some significant changes in the geometry, but only a small change in the imaginary mode to 979 cm-1 (entry 3).
Changing the functional from B3LYP to MN15L (entry 4) significantly reduces the imaginary mode value and here the effect of improving the basis set quality (entry 5) is large, reducing the imaginary transition state mode to 497 cm-1 Entry 6 shows the values for the r2scan-3c functional discussed in an earlier post,[8] revealing a transition state mode similar to the others. The free energy barriers range from 27.0 kcal/mol quoted in the article[1] down to 18.3 (entry 3) with the r2scan-3c functional being rather higher. Given that this reaction proceeds at temperatures of 253 – 298K, one might expect a barrier closer to the lower end of this range rather than the reported computed value of 27.0 kcal/mol and in this regard, the value for the r2scan-3c functional seems quite reasonable.
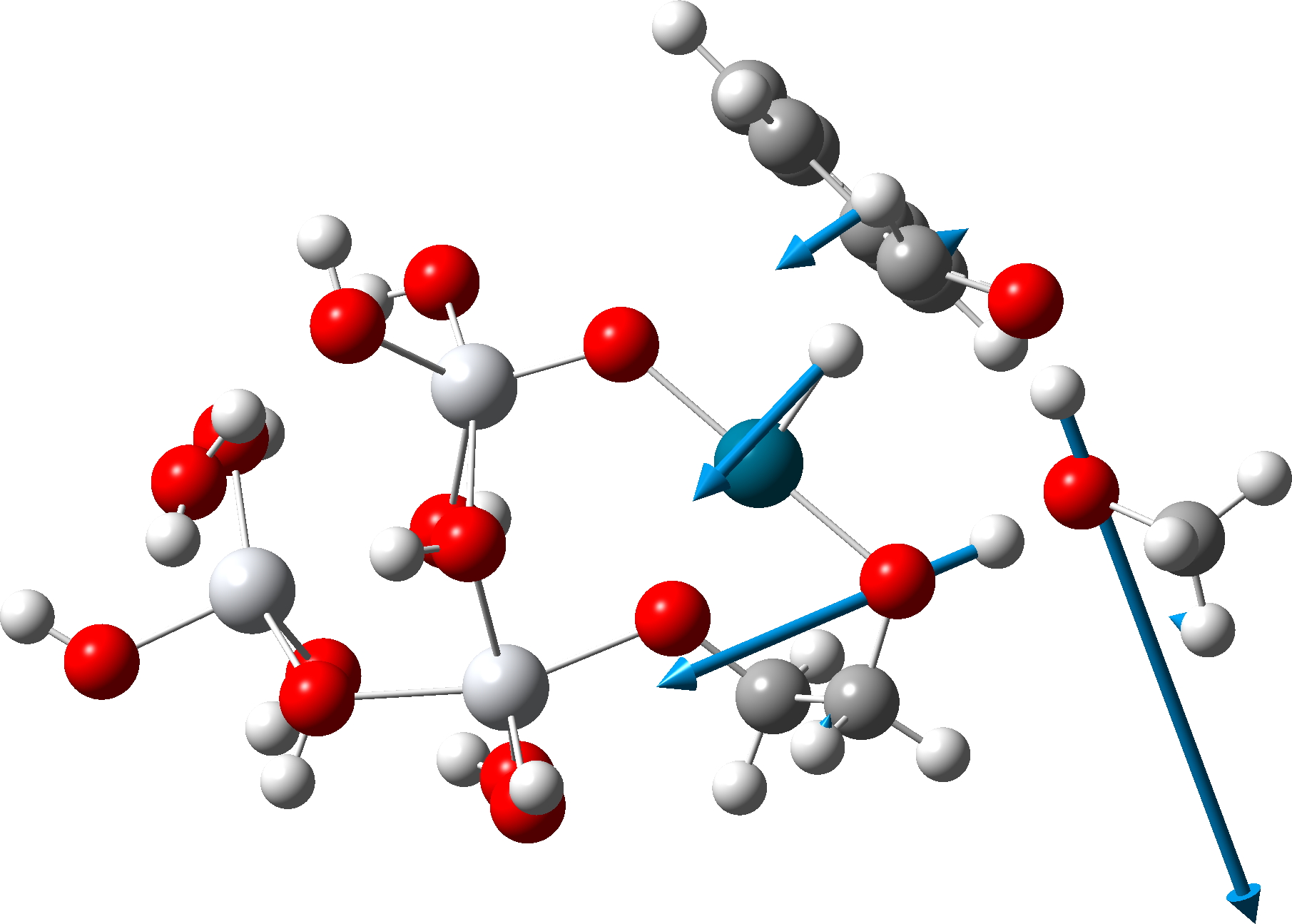
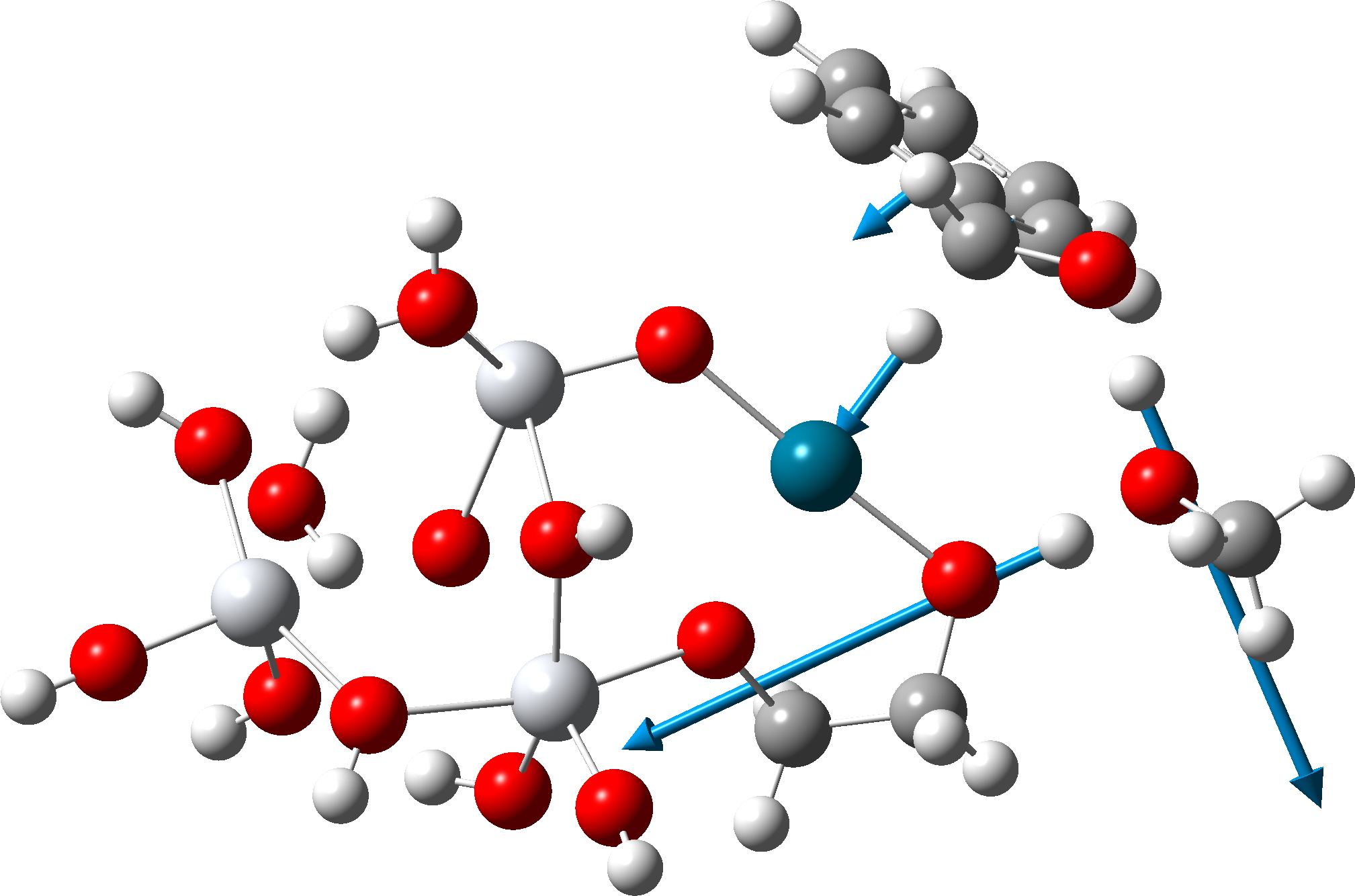
The transition state mode vibrational vectors are quite similar (entries 3 and 6 shown respectively below), indicating that the PD-H-C and adjacent O-H-O contributions are quite similar, whilst the final third transfer has a smaller contribution. This shows that the three transfers are not exactly synchronous, and hence any tunnelling contributions for the three transfers are unlikely to be the same.
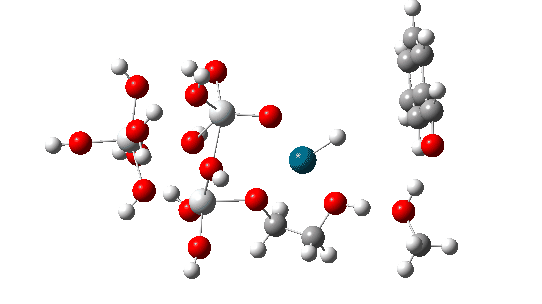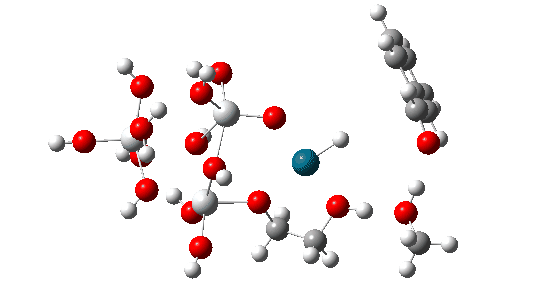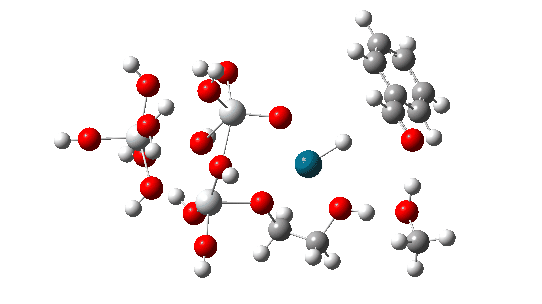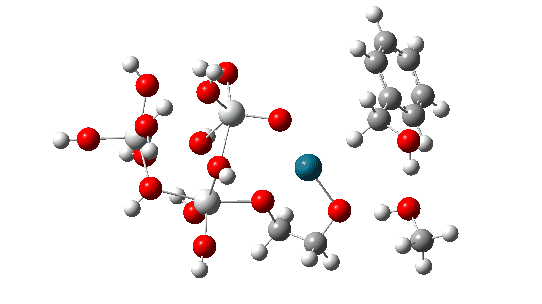
| row | Method | rPd-H | rH-C | rO1-H, Å | rH-O2 | rH-O2 | rH-O3 | α Pd-H-C, ° | νi | ΔG |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | B3LYP/6-311+G(d,p)/Lanl2dz gas phase† |
1.694 | 1.297 | 1.269 | 1.158 | 1.153 | 1.273 | 166.1 | 1306 | 27.0 |
| 2 | B3LYP/6-311+G(d,p)/Lanl2dz gas phase‡[9],[10] |
1.677 | 1.303 | 1.258 | 1.151 | 1.141 | 1.278 | 177.1 | 940 | 18.6 |
| 3 | B3LYP/6-311+G(d,p)/Lanl2dz/ | 1.736 | 1.241 | 1.295 | 1.132 | 1.181 | 1.229 | 177.2 | 979 | 18.3 |
| 4 | MN15L/6-311+G(d,p)/Lanl2dz/ | 1.703 | 1.283 | 1.360 | 1.096 | 1.082 | 1.383 | 143.8 | 497 | 25.8 |
| 5 | MN15L/Def2-TZVPP/ SCRF=methanol |
1.633 | 1.363 | 1.306 | 1.120 | 1.073 | 1.405 | 151.3 | 935 | 28.0 |
| 6 | r2scan-3c/Def2-mTZVPP/
SCRF=methanol |
1.639 | 1.360 | 1.212 | 1.199 | 1.129 | 1.290 | 136.7 | 985 | 21.3 |
Before a transition state model can be used to infer the KIE for isotopic substitution, it has to be tested against e.g. crystal structures and variation in more accurate basis sets and density functionals. The geometry of the transition state should also be optimised to high accuracy. Whether the KIE reported (~2440) would survive modelling at these more accurate levels remains to be seen. Or indeed whether such an exceptionally high value is directly related to the synchrony of the three hydrogen transfer shown above (“triple hydrogen tunneling”).
♥The largest value I know of that has been claimed for a KIE is the phenomenal value of ~1016[14] †[1] SI Table S7 etc.
References
- Q. Wu, P. Liu, X. Zhang, C. Fan, Z. Chen, R. Qin, Y.Q. Gao, Y. Zhao, and N. Zheng, “Catalytic Hydrogenation Dominated by Concerted Hydrogen Tunneling at Room Temperature”, ACS Central Science, vol. 11, pp. 2180-2187, 2025.
- C. Di Bugno, M. Pasquali, P. Leoni, P. Sabatino, and D. Braga, “Oxidative addition of phenols to bis(tricyclohexylphosphine)palladium. Synthesis and structural characterization of trans-[Pd(PCy3)2(H)(OC6H5)].C6H5OH (1) and trans-[Pd(PCy3)2(H)(OC6F5)].C6F5OH (2)”, Inorganic Chemistry, vol. 28, pp. 1390-1394, 1989.
- E.M. Richards, L. Casarrubios, J.M. D’Oyley, H.S. Rzepa, A.J.P. White, K. Goldberg, F.W. Goldberg, J.A. Bull, and S. Díez‐González, “Bidentate NHC‐Containing Ligands for Copper Catalysed Synthesis of Functionalised Diaryl Ethers”, Advanced Synthesis & Catalysis, vol. 367, 2024.
- H.S. Yu, X. He, and D.G. Truhlar, “MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids”, Journal of Chemical Theory and Computation, vol. 12, pp. 1280-1293, 2016.
- D.C. Braddock, S. Lee, and H.S. Rzepa, “Modelling kinetic isotope effects for Swern oxidation using DFT-based transition state theory”, Digital Discovery, vol. 3, pp. 1496-1508, 2024.
- H. Rzepa, “Triple-H”, 2025.
- B.P. Pritchard, D. Altarawy, B. Didier, T.D. Gibson, and T.L. Windus, “New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community”, Journal of Chemical Information and Modeling, vol. 59, pp. 4814-4820, 2019.
- H. Rzepa, “Short B-H…H-O Interactions in crystal structures – a short DFT Exploration using B3LYP+D4 and r2scan-3c”, 2025.
- H. Rzepa, “Triple-H with methanol, B3LYP/6-311+G(d,p)/Lanl2dz, Gas phase Reactant G = -4277.126369”, 2025.
- H. Rzepa, “Triple-H with methanol, B3LYP/6-311+G(d,p)/Lanl2dz, Gas phase G = -4277.096955”, 2025.
- H. Rzepa, “Triple-H with methanol, B3LYP/6-311+G(d,p)/Lanl2dz, Gas phase G = -4277.096955 => SCRF=methanol G = -4277.142864 DG = -28.8”, 2025.
- H. Rzepa, “Triple-H with methanol, B3LYP/6-311+G(d,p)/Lanl2dz, scrf=methanol, Reactant G = -4277.172103”, 2025.
- H. Rzepa, “Triple-H with methanol, MN15L+G(d,p)/Lanl2dz, SCRF=methanol G = -4274.865566 DG = 25.78”, 2025.
- N.D. Aisyah, R.N. Fadilla, H.K. Dipojono, and F. Rusydi, “A Theoretical Study of Monodeuteriation Effect on the Rearrangement of Trans-HCOH to H 2 CO via Quantum Tunneling with DFT and WKB Approximation”, Procedia Engineering, vol. 170, pp. 119-123, 2017.
This entry was posted on Friday, November 21st, 2025 at 1:24 pm and is filed under Interesting chemistry. You can follow any responses to this entry through the RSS 2.0 feed.
You can leave a response, or trackback from your own site.