
In an earlier blog, I discussed[cite]10.59350/rzepa.28849[/cite] the curly arrows associated with the known dimerisation of nitrosobenzene, and how the N=N double bond (shown in red below) forms in a single concerted process.

One of the properties of this molecule is that the equilibrium between the monomer and dimer can be detected[cite]10.1002/mrc.1260251118[/cite], with significant concentrations of the dimer observed below 10°C. This dimer can even be crystalised, with around 20 well defined crystal structures known for the dimeric structure in the current version of the CSD crystal structure dataset. Nitrosobenzene dimer itself forms a cis isomer, but others are known as trans (see below).
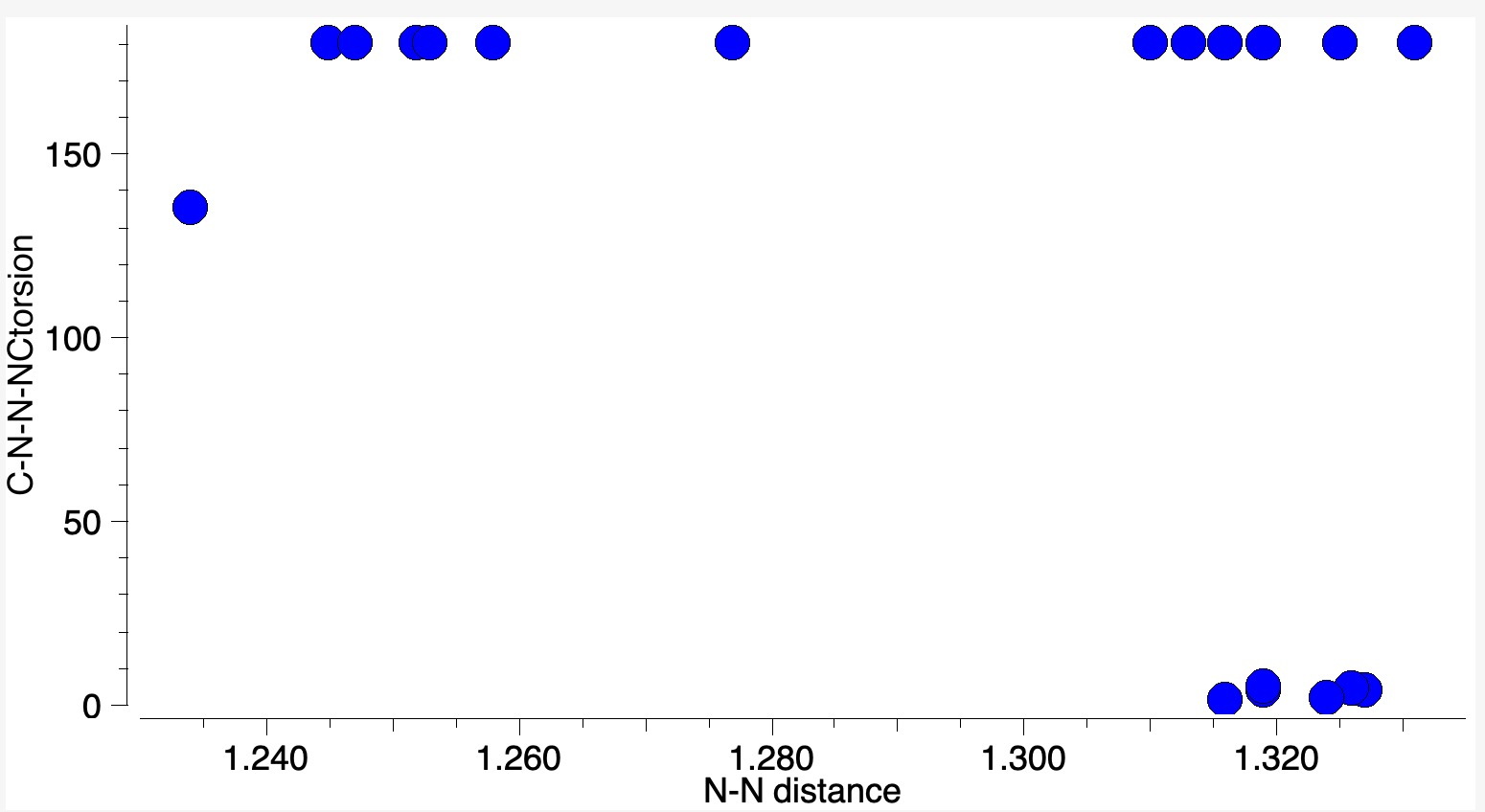
This detectable equilibrium means that the formal bond dissociation energy of that N=N bond must be very low – close to zero. This makes it an unusually weak double bond! Let’s explore how unusual by adopting a technique for analysing the energies in the molecule known as Natural Energy Decomposition Analysis or NEDA[cite]https://doi.org/10.1063/1.466432[/cite] (there are several other well-used methods for this, but I will concentrate on this one in this post at least). To explain what it is, I will paraphrase the NBO7 manual:
Natural energy decomposition analysis is an energy partitioning procedure for molecular interactions with contributions from Electrical interaction (EL), charge transfer (CT), and core repulsion (CORE) terms as evaluated for self-consistent field (SCF) wavefunctions.
- The electrical term EL = ES + POL + SE arises from classical electrostatic (ES) and polarization interactions (POL+SE). SE is the linear response self energy (energy penalty) of polarization.
- The CORE contribution CORE = EX + DEF − SE results principally from intermolecular exchange interactions (EX) and deformation (DEF), where the latter is the energy cost to distort a fragment wavefunction in the field of all other fragments of the complex. For DFT-based analysis, EX is replaced by the exchange-correlation interaction (XC).
- The total interaction energy is then given by
- ΔE = EL + CT + CORE
So now for some calculations[cite]10.14469/hpc/15455[/cite]. To do this, one has to consider an appropriate reference state[cite]10.1021/acs.organomet.5b00429[/cite] for the two fragments of the molecule, in this case nitrosobenzene itself. This is expressed via a set of charge,multiplicity definitions for the supermolecule and all the fragments. For the nitrosobenzene dimer, two possibilities can be considered
- 0,1 0,1 0,1 (which defines singlet states for all three species)
- 0,1 0,3 0,-3 (which defines triplet states for the two fragments, with a “spin flip” for the second).
Firstly I will calculate ΔE (Z)-1,2-diphenylethene, which is a classical C=C double bond alkene.
- For the reference state 0,1 0,3 0,-3
Electrical (ES+POL+SE) : -8691.975 Charge Transfer (CT) : -809.587 Core (XC+DEF-SE) : 9327.995 ------------ Total Interaction (E) : -173.567 kcal/mol - For the reference state 0,1 0,1 0,1 (which represents two carbenes)
Electrical (ES+POL+SE) : -7878.192 Charge Transfer (CT) : -918.005 Core (XC+DEF-SE) : 8473.018 ------------ Total Interaction (E) : -323.179 kcal/mol
So this classical C=C double bond partitions into two interacting triplet carbenes, with a spin flip to align their interaction. Now for nitrosobenzene.
- For the reference state 0,1 0,1 0,1 (which represents two nitrosobenzenes each with a lone pair of electrons)
Electrical (ES+POL+SE) : -18230.176 Charge Transfer (CT) : -818.925 Core (XC+DEF-SE) : 19021.537 ------------ Total Interaction (E) : -27.564 kcal/mol - For the reference state 0,1 0,3 0,-3
Electrical (ES+POL+SE) : -17567.592 Charge Transfer (CT) : -677.676 Core (XC+DEF-SE) : 18197.205 ------------ Total Interaction (E) : -48.063 kcal/mol
This shows completely different behaviour for the nitrosobenzene dimer and (effectively) the phenyl carbene dimer, with a different reference state for the two species. The electrical and charge transfer terms for the former are much larger than for the latter and this analysis does indeed conform the supposition made at the start that the N=N bond in nitrosobenzene dimer is indeed very unusual and very weak! Perhaps the weakest double bond known? If there are other candidates, I would love to hear about them!
Finally, I note that the relatively low NEDA energy for a triplet reference state for the nitrosobenzene dimer also matches with the observation made previously[cite]10.59350/rzepa.28849[/cite] that open shell (biradical) wavefunctions are needed to describe the curly arrows for the process.
Energy decomposition analysis is a good tool to have in one’s toolbox for analysing molecular behaviour and no doubt I will use it more in the future! Next, tetra-t-butylethene!
This post has DOI: 10.59350/rzepa.29383
Related
You can leave a response, or trackback from your own site.
3 Comments
https://shorturl.fm/MlJrD
https://shorturl.fm/Dm9We
https://shorturl.fm/Iw4u6